

Los -no tan nuevos- retos cuando se habla del virus del PRRS
No es ninguna novedad que el virus PRRS presenta importantes características que contribuyen a la constante reaparición de la enfermedad en las granjas porcinas, lo que provoca considerables problemas sanitarios en los animales y pérdidas económicas. Una de estas características es su rápida capacidad para mutar y volverse potencialmente más grave y/o incluso infectar animales previamente “inmunes” a variantes diferentes de PRRS. Por esta razón, con el fin de conocer el "tipo" de vPRRS presente en una granja – ya sea el virus de un nuevo brote o un virus “residente”- frecuentemente se analiza el genoma del vPRRS.
Esta tarea se realiza normalmente secuenciando una parte importante del vPRRS llamada gen ORF5; para entender dónde encaja evolutivamente el virus y cómo se compara con virus detectados anteriormente en la granja o en las proximidades/región.

La información que aporta la secuenciación del vPRRS se ha usado ampliamente en el campo para:
- entender qué virus está causando problemas
- para orientarse sobre el origen del brote
- para diferenciar entre cepas vacunales y cepas “residentes”/recién introducidas
- para la elección de vacunas, etc.
Recientemente, la clasificación por “linaje” se ha utilizado cada vez más para clasificar los virus. Este método se desarrolló alrededor de 2010 (Shi et al., 2010), cuando los vPRRS se dividieron en 9 linajes distintos; y se precisó aún más cuando se subdividieron en sublinajes.
Una verdad incómoda: ¿es suficiente secuenciar una muestra?
La secuenciación de PRRS no suele ser barata, por lo que se realiza generalmente en “situaciones especiales” y no de forma rutinaria (p.ej. durante nuevos brotes, después de casos de inoculación del virus, etc.). Además, en estas situaciones, en la gran mayoría de los casos estudiados para diagnóstico, sólo se usa una muestra o pool de muestras para la secuenciación, de las muchas que suelen tomarse en la granja.
La repercusión es que, en muchos casos, los veterinarios, productores y demás personal relacionado con la sanidad animal suelen utilizar la información de una única secuencia para sacar conclusiones importantes respecto al origen del virus (especialmente en nuevos brotes), orientar las investigaciones en los brotes e informar sobre futuras intervenciones.
En nuestro estudio, queríamos confirmar que, si secuenciábamos múltiples muestras recogidas en el mismo día, todas proporcionarían la misma secuencia, o secuencias que serían “lo suficientemente cercanas” para ser consideradas al menos del mismo linaje de virus. También queríamos intentar la secuenciación del genoma completo (WGS, por sus siglas en inglés) junto con la secuenciación del ORF5 para ver si obteníamos más información. WGS podría tener una ventaja, ya que proporciona información sobre todo el virus, mientras que la ORF5 se ha utilizado tradicionalmente, pero solo nos cuenta una pieza de la historia. Sin embargo, como todavía es un desarrollo relativamente reciente y puede ser caro (en Estados Unidos cuesta aproximadamente tres veces más que la secuenciación del ORF5), aún estamos aprendiendo a interpretarla y, nuestra “base de datos” para establecer comparaciones es mucho más pequeña respecto a la de ORF5.
Sospechábamos que, dada la rápida tasa de mutación del PRRS y el gran tamaño de las granjas porcinas modernas, podríamos encontrar múltiples linajes de PRRS dentro de un evento de muestreo, lo que sería preocupante. En la Fig. 1 se muestra un resumen gráfico del razonamiento de nuestro estudio.
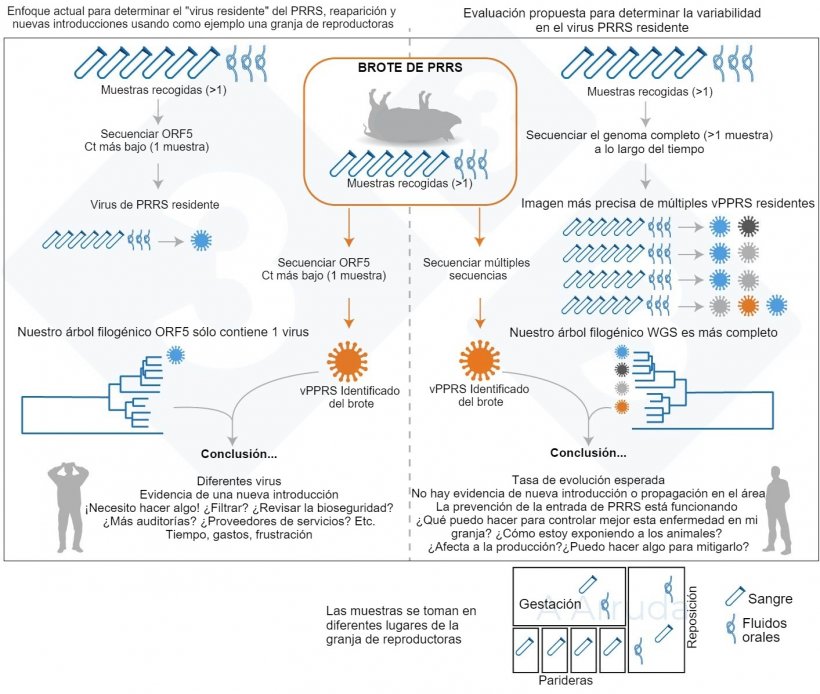
Investigando cuántas cepas de PRRS podemos encontrar en nuestras granjas
Incluimos cinco granjas en el estudio, 3 granjas de reproductoras y 2 granjas de engorde; y recogimos muestras mensualmente durante aproximadamente un año utilizando diferentes tipos de muestras (raspados de tonsilas, fluidos orales, fluidos del procesado). Siempre tomamos muestras de los mismos lugares en la nave que estaban repartidos espacialmente por las instalaciones para intentar obtener la mayor representación posible de todo el sitio. Recogimos hasta 16 muestras por sitio cada mes, y todas ellas se analizaron usando PCR cuantitativa; a continuación, se secuenció el ORF5 en las muestras positivas.
¿Qué hemos averiguado?
Nuestro hallazgo más importante fue que, en condiciones de campo, fuimos capaces de detectar hasta tres linajes diferentes del vPRRS durante un único evento de muestreo en granjas de reproductoras y hasta dos en granjas de engorde (Figura 2).
Figura 2. Sublinajes de vPRRS encontrados en las granjas de estudio (1-5) a lo largo de la duración de nuestros eventos de muestreo (en meses) para los diferentes tipos de muestras.
| Evento de muestreo | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Granja | Tipo de muestra | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 1 | Fluido del procesado | L1H | L1H | ||||||||||
| Raspado de tonsilas | |||||||||||||
| 2 | Fluido del procesado | L1H | L1H | L1A | L1H | L1H | |||||||
| L1H | |||||||||||||
| L8† | |||||||||||||
| Raspado de tonsilas | L1H | ||||||||||||
| 3 | Fluido del procesado | L1H | L1H | L1H | L1H | ||||||||
| Raspado de tonsilas | |||||||||||||
| 4 | Fluidos orales | L5† | L1A | ||||||||||
| L5† | |||||||||||||
| Raspado de tonsilas | L1A | L5† | L5† | L1A | |||||||||
| L5† | |||||||||||||
| 5 | Fluidos orales | L1A | L1A | L5† | |||||||||
| Raspado de tonsilas | L1A | L1A | |||||||||||
También observamos que, en el caso de nuestro estudio, las muestras de fluidos orales fueron muy difíciles de secuenciar, mientras que las de fluidos del procesado fueron las más fáciles. Teniendo en cuenta que ambas son muestras "compuestas", esto podría explicarse por los valores Ct más altos, que indican una menor cantidad de virus en los fluidos orales en comparación con los fluidos del procesado. Intentamos realizar WGS, pero finalmente no pudimos obtener secuencias completas de todo el genoma de ninguna de nuestras muestras. Sospechamos que podría deberse a los tipos de muestras utilizadas, pero es necesario seguir investigando sobre este tema.
Estos hallazgos demuestran lo importante que es considerar la secuenciación de varias muestras a la hora de tomar decisiones importantes, aunque entendemos que esto conllevará mayores costes.
Agradecimientos
El National Pork Board ha financiado este proyecto. Nos gustaría dar las gracias a los veterinarios y productores que fueron fundamentales para la recogida de muestras.


")