

El virus de la influenza A (IAV) es uno de los patógenos respiratorios más importantes en porcino. Tiene un impacto sobre la mortalidad y causa pérdidas económicas significativas por el descenso de productividad y los costes asociados a las vacunaciones y el tratamiento. Además, debido a la susceptibilidad de los cerdos a infecciones con IAV de distintas especies, en concreto humanos, pueden emerger nuevos virus por reordenamiento genético que tengan implicaciones para la salud pública. En consecuencia, la comprensión de la diversidad genética de los virus circulantes en porcino puede identificar nuevos linajes virales y proporcionar un criterio en el que basar y mejorar las estrategias de intervención.
La diversidad genética del IAV porcino en los EEUU es producto de la deriva y cambio antigénico, junto con la introducción de IAV de otras especies en las poblaciones porcinas (Vincent, et al. 2008). En términos generales, hay co-circulación de tres subtipos dominantes (H1N1, H1N2 y H3N2) y cuatro linajes genéticos principales descritos según el origen de los segmentos génicos. El linaje clásico porcino H1 se originó a partir de la pandemia de la gripe española de 1918. El segundo linaje se detectó a finales de los 1990s a partir de la hemaglutinina (HA) y la neuraminidasa (NA) derivadas de la gripe humana estacional H3N2. Este linaje era un virus nuevo reordenado, que contenía segmentos génicos HA, NA y PB1 derivados de la gripe humana estacional H3N2, segmentos génicos PB2 y PA de influenza aviar y segmentos génicos NP, M y NS de influenza porcina clásica H1N1, por lo que se denominó virus "reordenado triple". Estos virus se reordenaron a su vez con virus clásicos H1N1 para adquirir la HA y/o la NA, resultando en nuevos tipos genéticos de virus H1N1 y H1N2. La constelación de genes internos de la reordenación triple (TRIG) permaneció relativamente consistente con los orígenes porcinos (genes M, NP y NS), aviares (genes PB2 y PA) y humanos (PB1) de los virus de la influenza. El tercer linaje se produjo a partir de infecciones repetidas del virus humano estacional H1 IAV en cerdos (lo que se conoce como "spillover", o evento en el cual un patógeno propio de una especie salta a otra especie) a principios del siglo XXI, creando dos linajes distintos de virus humanos estacionales H1N1 y H1N2. El cuarto y más reciente linaje, representa un "spillover" del virus humano estacional H3N2 que tuvo lugar en 2010-11. Dentro de estos cuatro linajes principales de HA, el virus sigue evolucionando, formando nuevos clados genéticos (figura 1).

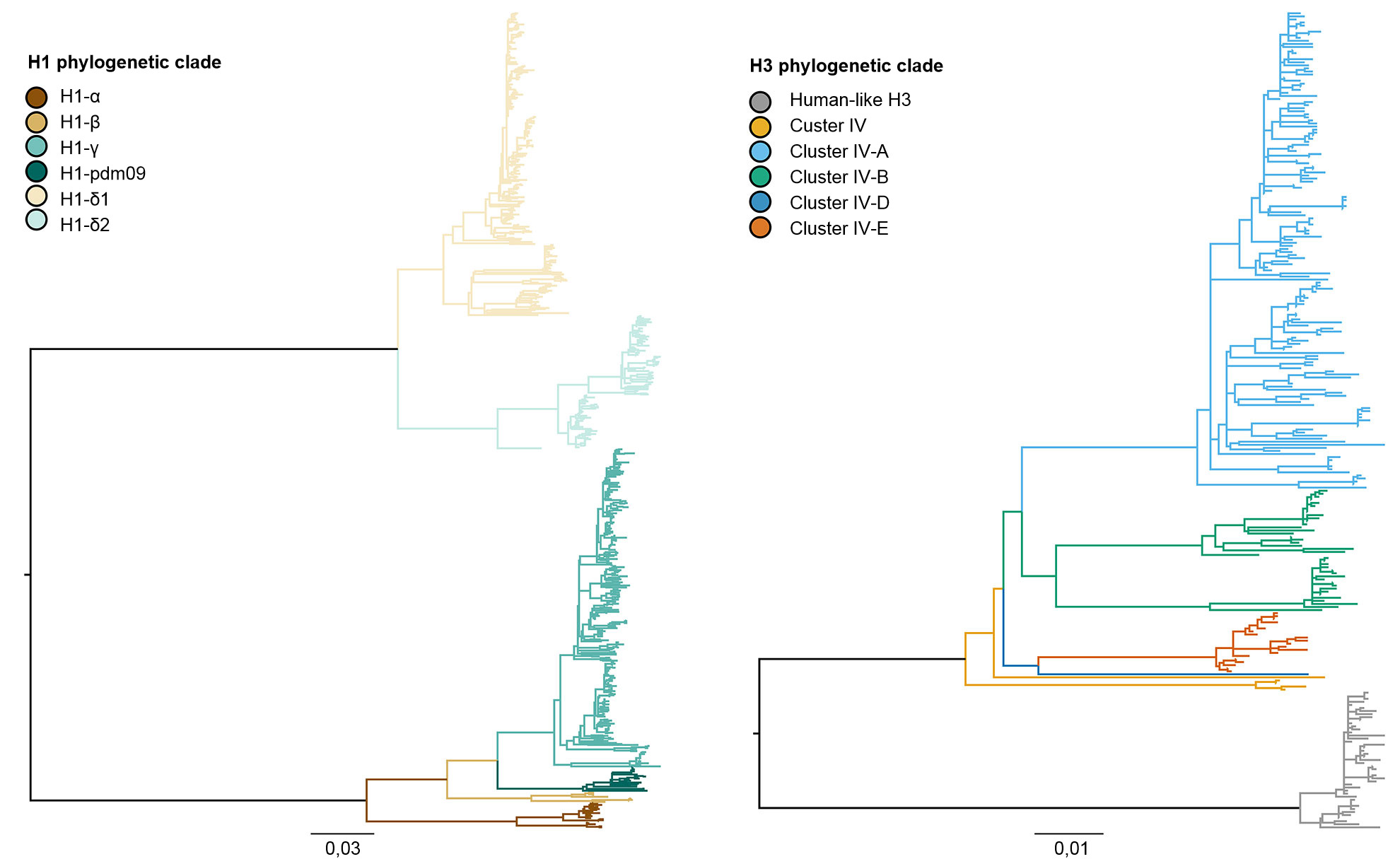
Figura 1. Árbol filogenético que describe las relaciones genéticas entre secuencias génicas de la hemaglutinina de la influenza A porcina H1 y H3 de 2015 generadas usando métodos de máxima verosimilitud. Las ramificaciones en color representan designaciones de clado. Las longitudes de las ramificaciones están dibujadas a escala, y la barra de escala indica el número de sustituciones de nucleótidos por sitio.
En un esfuerzo para mejorar la comprensión de la diversidad genética del IAV porcino y monitorizar el virus pandémico 2009 H1N1 (H1N1pdm09) en porcino, en 2009 se puso en marcha un sistema nacional de vigilancia del departamento de agricultura de EEUU (USDA) que se implementó a través de la red nacional de laboratorios de sanidad animal (NAHLN). El sistema se basa en envíos anónimos de productores y veterinarios; sin embargo, el sistema adolece de una pobre información geográfica sobre la procedencia de las muestras. Los datos epidemiológicos recogidos incluyen el estado, el tipo de muestra, el motivo del envío, la edad, el tipo de ubicación, resultados del análisis y, si es aplicable, se secuencian los genes HA, NA y M y se depositan en la base de datos online de secuencias del Centro Nacional de Información Biotecnológica, GenBank (Korslund et al. 2012; Anderson et al. 2013). Estos datos, y la inversión continuada en el sistema, han proporcionado información sobre los patrones de propagación del IAV, su diversidad genética a lo largo del año y sobre la dinámica de la evolución del IAV en Norteamérica desde 2010 hasta el presente (Anderson et al., 2013; Anderson et al., 2015; Lewis et al. 2014; Rajão et al. 2015).
Los tres subtipos (H1N1, H1N2 y H3N2) endémicos en la población porcina de EEUU han sido detectados cada año desde 2010 hasta la actualidad. Los subtipos H1N1 y H1N2 se detectaron con frecuencias similares (~35%) durante los 5 años. El subtipo H3N2 representa ~30% de los virus identificados durante todo el periodo. Para describir la diversidad genética de los virus H1 se utiliza un sistema basado en letras del alfabeto griego para dividir los datos de clados de HA dentro de los linajes clásicos (α, β, γ, γ-2 y H1N1pdm09) o del linaje humano (δ-1, δ-2). De un modo parecido, los genes del clúster IV de H3 se separan en 6 clados genéticos (A a F) y un clado H3 tipo humano (Rajão et al., 2015; Kitikoon et al. 2013). Sin embargo, pese al potencial de circulación de los 14 clados genéticos, la mayoría de la diversidad de HA observada en las granjas porcinas de EEUU se limita a tres clados. Desde enero de 2015 a diciembre de 2015, el 43% de los aislados H1 pertenecían al clado γ del linaje clásico y el 37% se clasificaron como δ-1 del linaje humano estacional; el 20% restante de los virus H1 procedieron de los clados δ-2, α, H1N1pdm09 y β. De los 8 clados potenciales H3, el clúster IV-A representa la mayoría de IAV H3 porcinos circulantes en las granjas comerciales (el 61% de los virus H3 durante 2015), se identificó un número creciente de aislados como H3 tipo humano (del 5% en diciembre de 2014 al 25% en diciembre de 2015), mientras que el resto de aislados representan detecciones esporádicas de los clústers IV-B, IV-C, IV-D, IV-E.
En el contexto de la eficacia de los programas de vacunación, hay una preocupación renovada de que la neuraminadasa (NA) también pueda desempeñar un papel importante debido a su diversidad creciente (Sandbulte et al., 2016). Aunque esto complica la situación, la NA tiene muchos menos linajes en que la HA. La HA está emparejada con los genes N2 derivados de uno o dos linajes humanos estacionales H3N2 (ya sean de 1998 o de 2002: (Nelson et al., 2011)), genes N1 del linaje clásico porcino, o genes del linaje estacional humano H1N1 (Anderson et al., 2013). En el actual IAV porcino circulante, el N1 pertenece, normalmente, al linaje clásico (94%) y el N2 al linaje 2002 (83%), mientras que el linaje 1998 representa, pese a ser detectado de un modo consistente, un componente pequeño de los aislados IAV.
Una segunda preocupación está relacionada con los patrones espaciales en la diversidad de IAV porcino. Los EEUU están divididos en cinco regiones en las que se reportan casos, basándose en los distritos veterinarios del USDA-APHIS (figura 2A). Hay sutiles e importantes diferencias entre regiones en cuanto a diversidad y abundancia de las muestras proporcionadas al sistema de vigilancia del USDA (figura 2B). Mientras que en las regiones 1 y 2 se detecta más H1N1, la región 3 tiene más H1N2 y la región 4 más H3N2. Pese a que el censo porcino es de 1,6 millones de cerdos (USDA-NASS, 2012), hay muy pocos datos de la región 5. La región 2 muestra la mayor diversidad en términos de distintos clados HA y NA observados y la mayoría de los aislados de IAV porcinos proceden de esta región. En general, la distribución de clados HA y NA sugieren que las decisiones regionales o locales sobre los componentes vacunales pueden ser importantes.
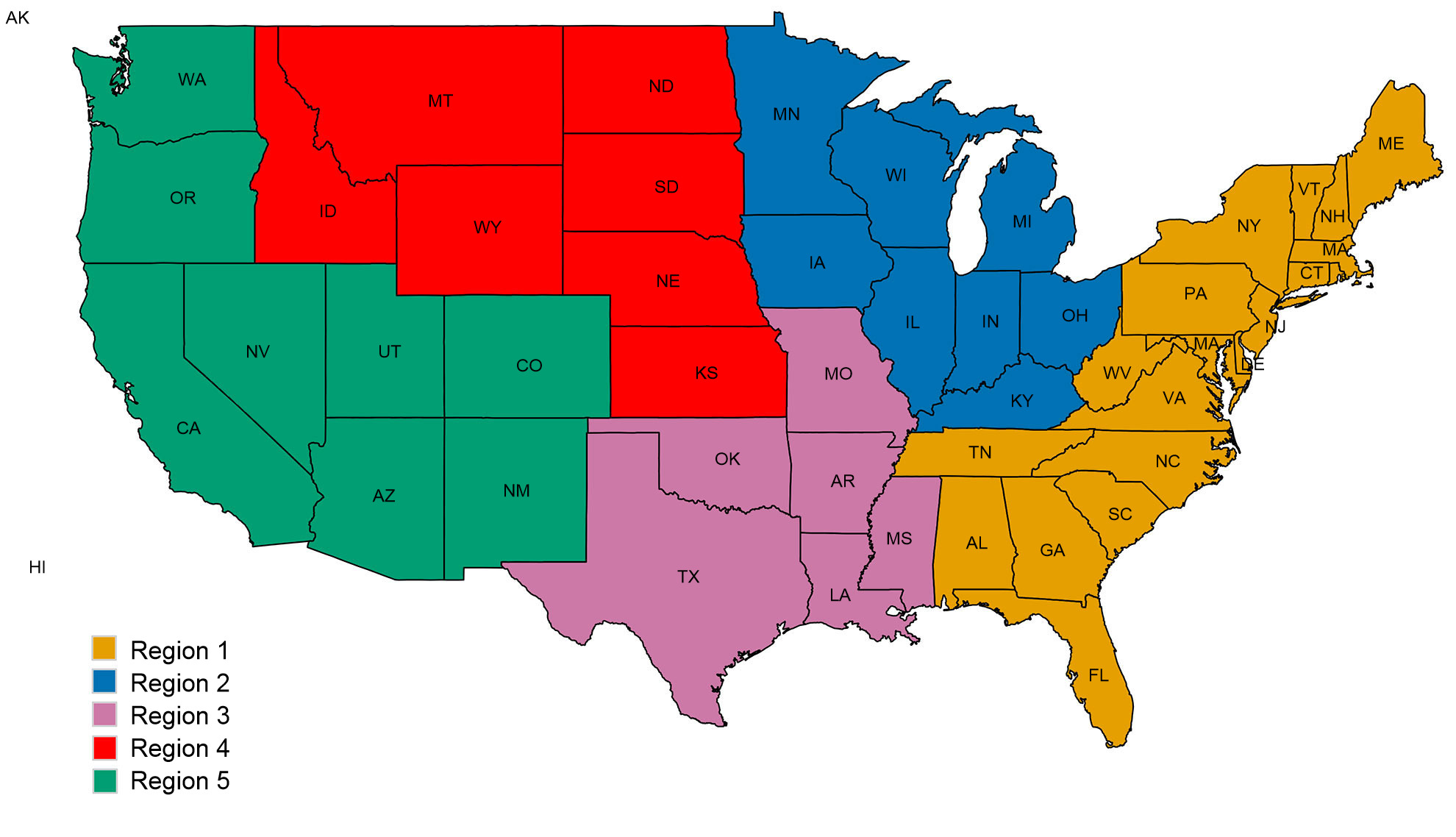
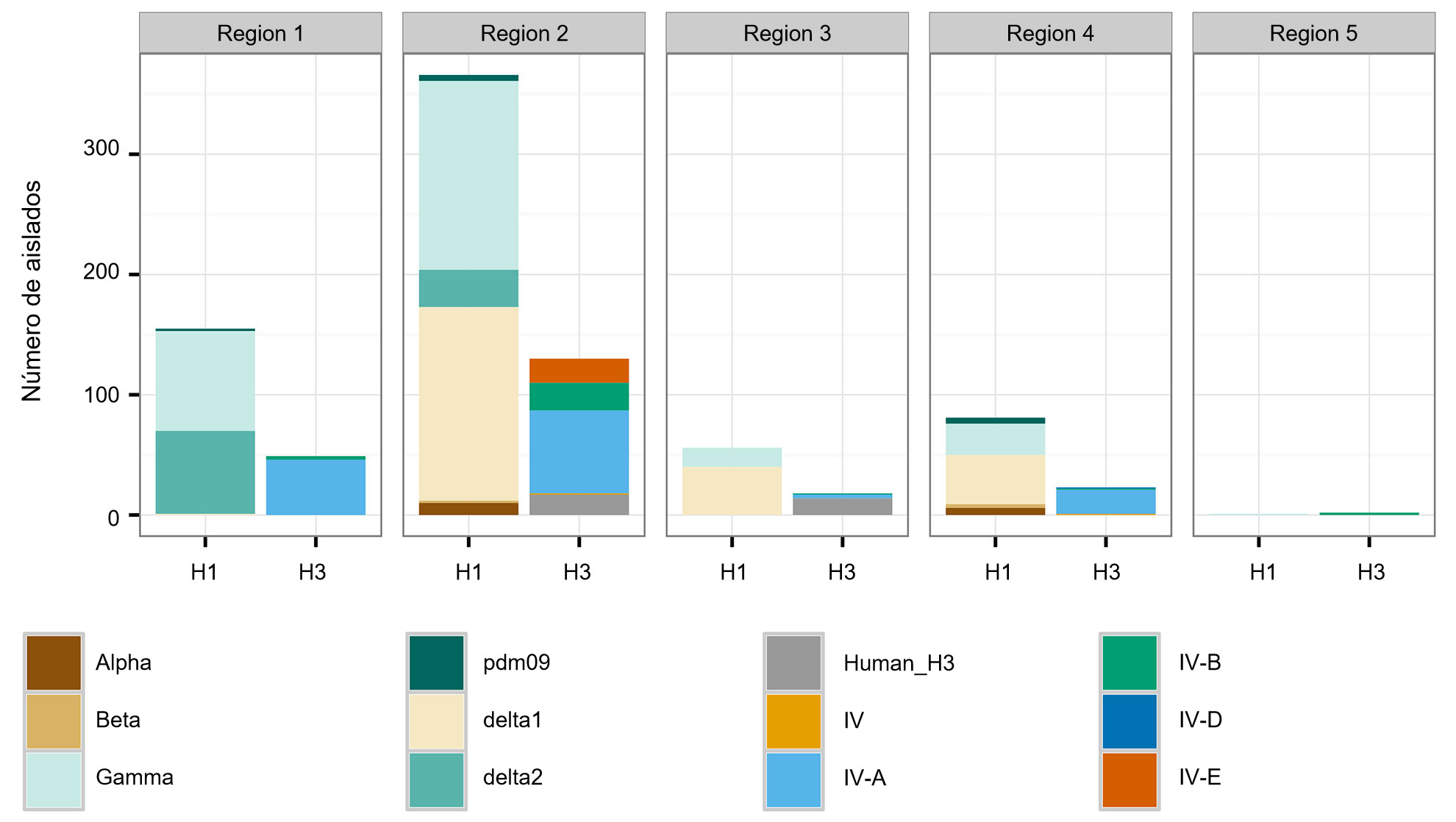
Figura 2. Regiones veterinarias del USDA-APHIS (A). Número de aislados de influenza porcina A recogidos en cada región durante 2015 y clasificados según clado filogenético y coloreado como en la figura 1 (B).
La diversidad genética del IAV porcino es un tema complejo a nivel regional y, especialmente a nivel global. En EEUU, hay 17 clados genéticos que han emergido y persistido a partir de eventos "spillover" entre hospedadores no porcinos (es decir, humanos) y los consiguientes procesos ecológicos y evolutivos. Pese a estar formada por distintos clados genéticos derivados de distintos episodios de "spillover" no porcinos, la diversidad genética es parecida en la población porcina mundial (por ejemplo Watson et al. 2015; Bahl et al. 2015; Vijaykrishna D et al. 2015). La genética del IAV porcino contemporáneo puede utilizarse para realizar estudios sobre evolución y diversidad antigénica y estos trabajos deberían ser llevados a cabo con datos actuales proporcionados por poblaciones regionales. Dicho conjunto de datos, con la implementación de una plataforma vacunal apropiada, proporcionaría los datos fundamentales para usar en la selección de cepas vacunales y proporcionaría información útil para las políticas de gestión de riesgos para la sanidad animal y pública.
