

Se incluyen en esta parte los datos relativos a la calidad del medicamento y sus ingredientes, el método de fabricación, controles y pruebas de estabilidad del producto. En azul señalamos dónde podemos encontrar referidos estos apartados en la ficha técnica del producto.
Consta de los siguientes apartados:

- Composición cuali-cuantitativa del medicamento: relación de las materias primas que constituyen el medicamento incluido el envase y su cantidad.
- Desarrollo farmacéutico: justificación y descripción de las pruebas que permiten seleccionar los excipientes utilizados y sus concentraciones en la composición final del medicamento. Algunos ejemplos de excipientes serían:
- Solventes (propilenglicol, agua)
- Tensioactivos (larilsulfato sódico)
- Reguladores de pH (ácido cítrico, carbonato sódico)
- Excipientes con acción LA (povidona, n-metilpirrolidona)
- Conservantes
De los dos puntos anteriores se obtienen el apartado 2 de la ficha técnica: Composición cualitativa y cuantitativa.
- Método de fabricación: Descripción paso por paso del proceso industrial de fabricación, validando mediante diferentes parámetros varios lotes del medicamento con el fin de comprobar le repetibilidad del resultado.

Productos en polvo y solución tras diferentes métodos de fabricación
- Materias primas: incluye a la sustancia activa o API (Active Pharmaceutical Ingredient), los excipientes y los materiales de acondicionamiento. Aquí se deben establecer los criterios de calidad que deberán cumplir todos los ingredientes utilizados, los mínimos los marca la Farmacopea Europea.
Debido a su importancia, vamos a ver más a fondo los requisitos necesarios para las sustancias activas.
- Se debe garantizar que el fabricante elabora la sustancia activa de acuerdo con las Buenas Prácticas de Fabricación (BPF), más conocidas por sus siglas en inglés GMP (Good Manufacturing Practice).
El fabricante del API, asimismo, debe aportar un Drug Master File o información detallada sobre la fabricación de la sustancia activa: reactivos, vías de síntesis, purificación, métodos de análisis, estabilidad, etc. La aparición de numerosos fabricantes de APIs en países emergentes, sobre todo localizados en Asia, que fabrican APIs de elevada calidad y a precios muy inferiores a los de los fabricantes tradicionales europeos, junto al boom de los genéricos, ha propiciado que los laboratorios busquen APIs más baratos en esos países para poder ofrecer precios competitivos. De hecho hay materias primas que prácticamente sólo se fabrican en China o India. La fuerte competencia existente en el sector farmacéutico y los ajustadísimos márgenes en los que se mueven algunos productos hace que APIs que no cumplen los requisitos exigidos de calidad de la Farmacopea Europea y que resultan más económicos puedan entrar en el mercado, lo cual supone un fraude.
Además de los criterios de calidad marcados por la Farmacopea Europea el fabricante puede ser más exigente con el fin de mejorar la biodisponibilidad del medicamento, por ejemplo utilizando materia prima micronizada.
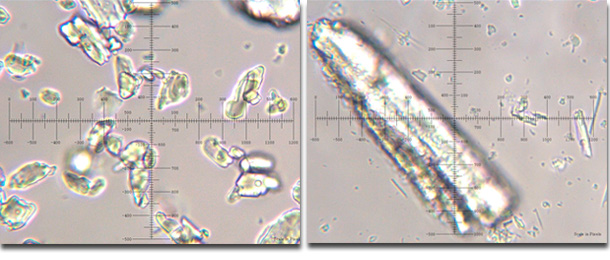
Ac. acetilsalicílico micronizado y no micronizado
- Control producto acabado: Se describen los criterios de calidad o especificaciones que debe de cumplir el medicamento junto a la descripción y validación de los métodos analíticos utilizados para ello: análisis y cuantificación de la sustancia activa, de los productos de degradación y las impurezas, de los conservantes si los hay y de las características físico-químicas y microbiológicas importantes dependiendo de la forma farmacéutica.
- Estabilidad: se aportan datos tanto de la estabilidad del principio activo como del producto acabado. Los primeros suele aportarlos el fabricante del API; los segundos el laboratorio que fabrica el medicamento.
La estabilidad del medicamento se suele realizar con 3 lotes diferentes almacenados a 25ºC y 60% de humedad relativa, del cual se van realizando análisis completos periódicamente a lo largo del tiempo. Suelen durar entre 3 y 5 años, que es el máximo periodo de validez aceptado por las autoridades. También se hacen estudios “acelerados” en los que el medicamento se somete a condiciones de temperatura y humedad elevados (40ºC y 75%h.r.). De aquí se obtienen el período de caducidad del producto y las condiciones de conservación requeridas.
Otros estudios de estabilidad que se realizan son los del medicamento una vez se abre el envase que lo contiene y si procede, del medicamento una vez reconstituido, disuelto en agua o mezclado con el pienso. De estos estudios se obtienen:
- La caducidad del producto una vez abierto el envase.
- Tiempo de renovación de la solución medicamentosa en los productos solubles.

Diferencias de estabilidad de dos productos una vez disueltos