

Wie werden ASP-Viren klassifiziert?
Das Virus der Afrikanischen Schweinepest (ASPV bzw. ASFV) wurde anhand der Sequenz des B646L-Gens, das für das p72-Protein – den Hauptbestandteil des Virus – kodiert, in verschiedene Genotypen eingeteilt. Basierend auf dieser Sequenz wurden bis zu 24 verschiedene Genotypen beschrieben, die auf dem afrikanischen Kontinent zirkulieren.
Außerhalb Afrikas wurden bislang nur die folgenden zirkulierenden Genotypen gemeldet:

- Genotyp I: derzeit in Asien verbreitet und für die Pandemie der 1960er Jahre verantwortlich
- Genotyp II: vorherrschender Genotyp der aktuellen Pandemie, die 2007 in Georgien ihren Ausgang nahm
Das europäische Referenzlabor hat innerhalb von Genotyp II eine zusätzliche Unterteilung in genetische Gruppen vorgeschlagen, basierend auf Unterschieden in den Sequenzen von sechs variablen Regionen des Virusgenoms.
| CVR | IGR I73R/I329L | O174L |
| K145R | IGR MGF505 9R/10R | IGR I329L-I215L |
Auf Grundlage der in diesen Regionen festgestellten Unterschiede wurden insgesamt 28 genetische Gruppen beschrieben, wobei die genetische Gruppe #1 durch den Stamm „Georgia 2007“ repräsentiert wird.
Die vorgeschlagene Nomenklatur für den in Barcelona nachgewiesenen Stamm lautet SP25WB2611, abgekürzt Sp25.
Der in Barcelona gefundene Stamm (Sp25) unterscheidet sich von den bisher bekannten Stämmen und wurde daher einer neuen genetischen Gruppe zugeordnet: #29.
Einige genetische Gruppen wurden ausschließlich bei Hausschweinen isoliert, andere nur bei Wildschweinen und wieder andere in beiden Wirten. Bestimmte genetische Gruppen, wie etwa #3 oder #19, treten in mehreren Ländern auf, während die meisten bislang nur in einem begrenzten geografischen Gebiet nachgewiesen wurden. Außerdem gibt es Regionen, in denen mehrere genetische Gruppen gleichzeitig zirkulieren, beispielsweise in der Russischen Föderation (#1, #2, #3, #4 und #7) sowie in Rumänien (#3, #7, #19, #21, #22 und #24).
Der Stamm Sp25 weist im Vergleich zum Stamm Georgia 2007 eine große Deletion von etwa 10 kb am linken Ende des Genoms sowie insgesamt 27 SNPs (Single Nucleotide Polymorphisms, d. h. Punktmutationen, bei denen ein einzelnes Nukleotid in der Virussequenz verändert wird) auf.
Wie ist die räumliche und zeitliche Verteilung der in der GenBank verfügbaren Sequenzen, mit denen die neuen Stämme verglichen werden?
Obwohl seit 2007 zahlreiche ASPV-Stämme in Europa isoliert wurden, ist die Zahl der verfügbaren Sequenzen im Vergleich dazu sehr gering.
Das Wissen über die geografische Verteilung genetischer Gruppen hängt von der Anzahl der Proben ab, die in den einzelnen Ländern sequenziert und veröffentlicht werden. In vielen Ländern, in denen die Afrikanische Schweinepest auftritt, werden die Ergebnisse jedoch weder sequenziert noch veröffentlicht.
Die am IRB und am BSC in Barcelona durchgeführten Sequenzanalysen des Stammes Sp25 belegen dessen phylogenetische Nähe zu Stämmen, die nach der Einschleppung von „Georgia 2007“ nach Europa bei Ausbrüchen in europäischen Ländern isoliert wurden (Abb. 1). Der Stamm Sp25 gehört zu einer Linie, die sich sehr früh abgespalten hat, und unterscheidet sich daher deutlich von den Stämmen, die die jüngsten Ausbrüche in Mittel- und Westeuropa verursachen. Wie das Baumdiagramm zeigt, ist diese Sequenz enger mit Viren verwandt, die 2019 in Russland nachgewiesen wurden, als mit den in Mittel- und Westeuropa isolierten Stämmen. Dennoch unterscheidet sie sich weiterhin deutlich von sämtlichen Stämmen, deren vollständiges Genom bislang bestimmt wurde, einschließlich der in Russland isolierten Stämme. Diese Unterschiede betreffen die erwähnte Deletion sowie mehr als 20 SNPs.

Das ASPV gilt als ein sehr großes und sehr stabiles DNA-Virus. Wie viele Veränderungen muss ein ASP-Isolat aufweisen, damit es als ein anderer Stamm betrachtet wird?
Im Rahmen der ASPV-Überwachung in Europa geht das Europäische Referenzlabor davon aus, dass ein Virusisolat als ein anderer Stamm betrachtet wird, wenn es einer neuen genetischen Gruppe zugeordnet wird.
Die Anzahl der nachgewiesenen SNPs gibt Aufschluss über die zeitliche Distanz eines Stammes vom „ursprünglichen“ Georgia-2007-Stamm. Das ASPV ist ein doppelsträngiges DNA-Virus und daher sehr stabil. Angesichts der niedrigen Mutationsrate des Virus müssen sich die verschiedenen SNPs, die Sp25 aufweist, über einen längeren Zeitraum seit seiner Abspaltung von Georgia 2007 angesammelt haben. Nach aktuellen Schätzungen könnte dieser Zeitraum bei etwa 10–15 Jahren evolutionärer Divergenz liegen (Schätzung von Dr. Toni Gabaldón, IRB).
Wenn es sich um ein derart stabiles Virus handelt, kann die bei dem im Rahmen des Ausbruchs in Barcelona gefundenen Stamm beobachtete Deletion von 10.000 Basenpaaren dann auf natürliche Weise entstehen?
Die natürliche Evolutionsdynamik des Virus kann genau solche genetischen Deletionen hervorrufen. Innerhalb des Genotyps II wurden bereits Deletionen in derselben Region mit unterschiedlicher Ausdehnung und unterschiedlicher Betroffenheit von Genen beschrieben, sowohl in Afrika (Ambagala et al., 2023) als auch in Europa (Zani et al., 2018; Torresi et al., 2025).
Diese Deletion wird bei einigen Impfstoffkandidaten verwendet. Bedeutet das, dass sie nicht natürlich auftreten kann?
Diese Art von Deletion im ASPV-Genom kommt in der Natur vor und kann auch im Labor erzeugt werden, um Impfstoffkandidaten zu entwickeln. Dabei ist zu beachten, dass heute im Rahmen der Entwicklung von Impfstoffkandidaten im Labor in den meisten Fällen ein exogener genetischer Marker in das Virusgenom eingefügt wird (aus technischen Gründen), der es ermöglicht, den Impfstoffkandidaten von dem ursprünglichen Virusstamm zu unterscheiden. Daher lassen sich Impfstoffkandidaten durch Sequenzierung leicht identifizieren. Der Stamm Sp25 weist keinen solchen exogenen Marker auf. Zudem werden die meisten Impfstoffkandidaten aus einer kleinen Gruppe sehr ähnlicher Laborstämme entwickelt, die untereinander eng verwandt sind und dem ersten Isolat des aktuellen Ausbruchs, Georgia 2007, sehr nahe stehen.
Kommt es häufig zu Rekombinationen zwischen verschiedenen ASP-Stämmen?
Damit solche Rekombinationen stattfinden können, muss eine Wirtszelle gleichzeitig mit zwei unterschiedlichen Stämmen infiziert sein. Dieses Phänomen ist weniger häufig als bei RNA-Viren mit segmentiertem Genom (wie es beim Influenzavirus der Fall ist), kommt beim ASPV jedoch durchaus vor. In China wurden Rekombinationen zwischen Genotyp I und II beschrieben (Zhao et al., 2023), und ein solches rekombinantes Virus hat sich anschließend auch in andere Länder (Russland und Vietnam) verbreitet. Logischerweise steigt die Wahrscheinlichkeit solcher Rekombinationen, je mehr unterschiedliche Stämme in einem Gebiet gleichzeitig zirkulieren. Das Muster der im Collserola-Stamm nachgewiesenen Mutationen erstreckt sich über das gesamte Genom und ist nicht auf eine begrenzte Region beschränkt, was einen Ursprung durch Rekombination unwahrscheinlich macht.
Es wird häufig gesagt, dass der in Barcelona nachgewiesene Stamm nicht besonders virulent sei. Ist das wirklich so?
Die im Stamm Sp25 identifizierte Deletion könnte mit einer geringeren Virulenz assoziiert sein, wie dies bereits bei anderen Stämmen mit Deletionen in dieser Region nachgewiesen wurde. Allerdings gibt es auch Beispiele hochvirulenter Stämme mit ähnlichen Deletionen, sodass kein eindeutiger Zusammenhang besteht. Die Virulenz des Stammes Sp25 kann daher erst abschließend beurteilt werden, wenn unter kontrollierten Bedingungen experimentelle Infektionsstudien an Schweinen oder Wildschweinen durchgeführt werden.
Die einzige derzeit gesicherte Erkenntnis ist, dass es sich um einen für Wildschweine tödlichen Stamm handelt und dass das beobachtete Krankheitsbild mit dem vergleichbar ist, das bei anderen europäischen Ausbrüchen beschrieben wurde, die durch als hochvirulent eingestufte Stämme verursacht wurden.
Abb. 2 zeigt eine zeitliche Rekonstruktion der Wildschweinfälle im Rahmen des aktuellen ASP-Ausbruchs, wobei zu berücksichtigen ist, dass das Datum des Auffindens eines Kadavers in der Regel nicht mit dem tatsächlichen Todeszeitpunkt des Tieres übereinstimmt. In jedem Fall wird ein plausibler Zeitrahmen für den Tod des Wildschweins angegeben, der anhand des Verwesungszustands des Kadavers geschätzt wurde. Auch wenn sich das genaue Datum der ersten Todesfälle nicht mit großer Genauigkeit bestimmen lässt, deuten die geschätzten Zeitintervalle der ersten Tierkadaver darauf hin, dass die mit dem Ausbruch verbundene Mortalität bereits Monate vor der Feststellung des ersten Falls einsetzte, wahrscheinlich etwa im September. Das ASPV zirkulierte daher zunächst unbemerkt, wahrscheinlich in geringem, aber anhaltendem Ausmaß, bis es schließlich nachgewiesen wurde. Die Anfangsphase des Ausbruchs, die sich langsam entwickelte, entspricht der Einschleppung und Etablierung des ASPV in der Wildschweinpopulation, gefolgt von einer epidemischen Phase, in der eine Veränderung der Kurvenneigung beobachtet werden kann. Der Verlauf dieses Ausbruchs unterscheidet sich nicht wesentlich von dem Muster, das bei jüngsten Ausbrüchen in Europa beobachtet wurde.
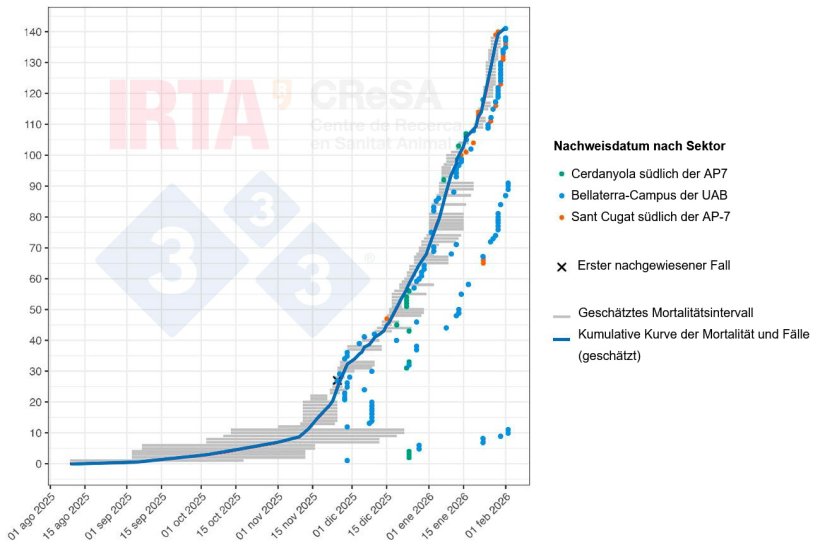
Abb. 2: Zeitliche Rekonstruktion der Mortalität und der ASP-Fälle bei Wildschweinen während des Ausbruchs in Collserola. Die grauen horizontalen Linien stellen die geschätzten Mortalitätsintervalle für jedes Tier dar. Die Punkte zeigen den Zeitpunkt, an dem der Kadaver oder das erkrankte Tier entdeckt wurde. Die Farbe jedes Punktes kennzeichnet den geografischen Sektor, in dem der Kadaver gefunden wurde (grün: Sektor Cerdanyola südlich der AP7; blau: Sektor Bellaterra-UAB-Campus; und orange: Sektor Sant Cugat südlich der AP7). Das Kreuz markiert den ersten nachgewiesenen Fall. Die blaue Linie stellt die kumulative Kurve der Mortalität und der ASP-Fälle dar.