

El virus del síndrome reproductivo y respiratorio porcino (vPRRS) es, junto con el virus de la peste porcina africana, uno de los patógenos de mayor impacto económico en la industria porcina mundial. La reciente aparición de cepas muy agresivas del vPRRS, como el vPRRS altamente patógeno (HP-vPRRS) en Asia, Rosalía en Europa y el L1C.5 en Norteamérica, ha encendido el debate sobre la necesidad de seguir mejorando el diagnóstico y control del vPRRS.
El uso de RT-PCR para detectar el material genético del vPRRS se utiliza de forma habitual en todo el mundo para analizar las pblaciones en busca del virus.

Un paso más allá de la detección del ARN es la secuenciación genética del vPRRS, que suele realizarse mediante la técnica de Sanger. A nivel mundial, la porción del virus más usada para la secuenciación del vPRRS es el ORF5, aunque algunos laboratorios generan informes con la secuenciación del ORF7.
El ORF5 representa aproximadamente el 4 % y el ORF7 el 2,4 % del genoma del vPRRS por lo que no proporcionan una cobertura genética completa de todo el genoma.
En los últimos años, ha habido un creciente interés en el uso de la secuenciación de nueva generación (NGS, Next Generation Secuencing) para la recuperación de genomas completos del vPRRS para investigaciones epidemiológicas dentro de una granja o sistema de producción (figura 1).
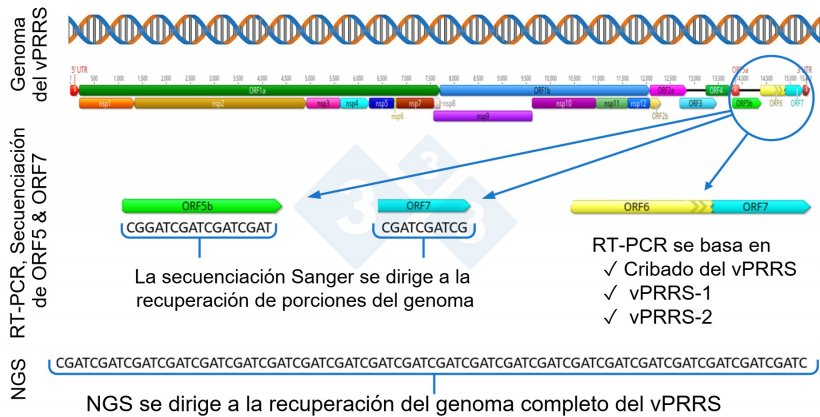
Figura 1: Representación esquemática de un genoma completo del vPRRS (GenBank U87392) y las regiones diana en las diferentes pruebas diagnósticas. En Estados Unidos, las pruebas RT-PCR para detectar virus vivos modificados (MLV) similares a los de la vacuna se dirigen a la región nsp2, y la secuenciación CLAMP para bloquear la amplificación de los virus de la vacuna MLV durante la secuenciación Sanger se dirige al gen ORF5.
Durante el proceso de replicación, el vPRRS sufre cambios genéticos y mutaciones, que potencialmente pueden conducir a la aparición de nuevas variantes del virus. El vPRRS es conocido por tener una alta tasa de mutaciones, aproximadamente del 0,5 al 1 % anual, distribuidas de forma desigual en diferentes regiones del genoma, distintos tipo de virus y linajes genéticos, lo que conduce a una evolución genética constante. En particular, la mutación y evolución genética del virus puede producirse en todos los genes, por lo que la secuenciación de sólo una parte del genoma, por ejemplo, ORF5 u ORF7, probablemente omita la detección de cambios ocurridos fuera de la región secuenciada (figura 1).
En este escenario, la NGS se convierte en una herramienta útil al proporcionar la oportunidad de recuperar un genoma completo del vPRRS para su uso en investigaciones epidemiológicas.
¿Cómo pueden los veterinarios y productores aprovechar la NGS?
Para maximizar la utilidad de la NGS, se deben considerar algunos puntos:
- Es importante que el veterinario y el laboratorio diagnóstico se impliquen desde el principio para alinearse respecto al diagnóstico diferencial, las expectativas y el enfoque de las pruebas.
- ¿Disponemos de una secuencia completa del genoma de una cepa de referencia de vPRRS de la granja o sistema de producción para comparar?
- Si no es así, utiliza la NGS en muestras positivas por RT-PCR para vPRRS con valores Ct bajos, es decir, idealmente <24, para recuperar un genoma completo como cepa de referencia de la granja. El valor Ct está inversamente relacionado con la cantidad de copias genéticas del virus presente en la muestra; es decir, cuanto menor sea Ct, mayor será la carga viral esperada y el éxito de la NGS;
- Una cepa de referencia permite la posterior comparación de genomas recuperados prospectivamente para comprender la evolución genética del vPRRS dentro de la granja, el flujo y el sistema.
- ¿Cuál es el objetivo de realizar una NGS?
- Si el objetivo es detectar la evolución del virus a nivel de genoma completo: se deben tomar muestras de pulmón y suero, ya que estas muestras tienen más probabilidades de recuperar un genoma completo.
- Si el objetivo es comprender la diversidad de virus en una granja, utiliza tipos de muestras poblacionales como fluidos de procesado, fluidos orales o muestras individuales agrupadas (pools), por ejemplo, las muestras de suero agrupadas son más propensas a recuperar múltiples virus si están presentes en la muestra.
- Las infecciones mixtas con dos o más virus PRRS en una granja son una realidad (figura 2a), y si los dos virus son muy similares, la NGS puede no recuperar un genoma completo;
- Si hay múltiples virus presentes en la muestra, la NGS puede recuperar fragmentos del genoma, conocidos como "contigs", de diferentes virus, que, al compararse con una cepa de referencia de la granja, pueden ayudar a discernir si hay múltiples virus presentes en la muestra (figura 2b);
- Por último, asegúrate de contar con alguien que te ayude con el análisis genético y las comparaciones, y coordina el tiempo de respuesta, ya que la NGS suele ser un proceso lento que puede tardar más de una semana en completarse.

Figura 2: A) Representación esquemática de una comparación de dos secuencias genómicas completas del vPRRS recuperadas de muestras de suero agrupadas 15:1 con Ct de 18,4. Se estableció un virus de referencia (color azul). El nivel de similitud de nucleótidos entre los virus está representado por números en los recuadros rojos. B) Representación esquemática de una comparación de dos secuencias genómicas del vPRRS recuperadas de muestras de suero agrupadas 5:1 con Ct de 19,5. Se estableció un virus como cepa de referencia (color azul). El nivel de similitud de nucleótidos entre los fragmentos del genoma recuperados del segundo virus está codificado por colores y también representado por números en los recuadros rojos. Los genes del genoma del vPRRS están representados en la parte superior de ambos paneles A y B. Los ORF individuales presentes en una secuencia del genoma completo del vPRRS están representados en la parte superior de ambos paneles A y B.
¿Qué tipo de información epidemiológica podemos obtener de la NGS?
Los resultados generados por la NGS pueden ayudar a resolver preguntas como:
- ¿El virus evolucionó mediante sustituciones aleatorias de nucleótidos en las mismas posiciones genómicas? ¿Cuánto ha cambiado el virus entre dos puntos temporales y en qué regiones del genoma, es decir, regiones ORF, ocurrieron estos cambios?
- ¿El virus evolucionó mediante inserciones o deleciones en su genoma?
- ¿Hubo una nueva introducción de un virus no relacionado en la granja o flujo?
- Aunque es menos probable pero posible, ¿el nuevo virus adquirió cambios en las regiones genómicas diana de la RT-PCR o de la secuenciación con cebadores/sondas Sanger, haciendo que las pruebas no detectaran el virus?
- ¿El virus experimentó una recombinación, es decir, adquirió algunas regiones genómicas de dos o más virus parentales?
La recombinación es un proceso natural de evolución del vPRRS y ocurre cuando dos vPRRS se replican en la misma célula, generando un tercer virus derivado. La recombinación se explorará más a fondo en un próximo artículo que publicaremos en junio de 2025, titulado "Las implicaciones del dilema de la recombinación del vPRRS".

