

Wirusowe DNA pełni rolę swoistego „odcisku palca”, a porównanie sekwencji uzyskanej z ogniska choroby z genomami referencyjnymi dostępnymi na świecie pozwala określić pokrewieństwo genetyczne pomiędzy poszczególnymi ogniskami.
Do klasyfikacji zastosowano ujednolicony schemat charakterystyki molekularnej opracowany przez Europejskie Laboratorium Referencyjne ds. Afrykańskiego Pomoru Świń (EURL-ASF, CISA-INIA/CSIC), który łączy podejście wielogenowe z sekwencjonowaniem całego genomu.

Charakterystyka wirusa ASF
Wirus ASF:
- posiada dwuniciowy genom DNA,
- jest wyjątkowo dużym i złożonym wirusem, którego genom obejmuje około:
- 170 000–194 000 nukleotydów,
- około 170 genów, w zależności od izolatu wirusa,
- wykazuje wysoki stopień konserwacji genetycznej, szczególnie w centralnej części genomu.
Jak klasyfikowane są wirusy ASF?
Poziom 1. Globalna klasyfikacja genotypowa wirusa ASF
Tradycyjnie wirus ASF klasyfikowany jest na podstawie sekwencji genu B646L, kodującego białko strukturalne p72. Na podstawie różnic występujących w tym genie możliwe jest wyodrębnienie pierwszego poziomu klasyfikacji wirusów, co pozwoliło dotychczas zidentyfikować 24 genotypy występujące na świecie.
Pomimo tej globalnej różnorodności, od momentu zawleczenia wirusa do Gruzji w 2007 roku z Afryki Wschodniej, to właśnie genotyp II jest głównym sprawcą obecnej pandemii ASF na świecie. Wirus ten rozprzestrzenił się w wielu krajach Europy, Azji i Oceanii, a w 2022 roku dotarł również do Dominikany i Haiti w obu Amerykach.
Obecnie genotyp II jest jedynym genotypem krążącym w Europie, zarówno w populacji dzików, jak i świń domowych.
Wirusy należące do genotypu II są genetycznie bardzo podobne, dlatego ten pierwszy poziom klasyfikacji nie pozwala na rozróżnianie blisko spokrewnionych wariantów ani na badanie pochodzenia i powiązań pomiędzy ogniskami choroby na poziomie regionalnym czy lokalnym.
Poziom 2. Podejście wielogenowe do różnicowania wirusów ASF w obrębie genotypu
Aby przezwyciężyć ograniczenia wynikające z wysokiego podobieństwa genetycznego wirusów należących do genotypu II, opracowano podejście wielogenowe oparte na sekwencjonowaniu 6 dodatkowych regionów genomu wirusa.
- Region międzygenowy (IGR) pomiędzy genami I73R i I329L,
- Centralny region zmienny (CVR) genu B602L,
- Gen O174L,
- Gen K145R,
- Region międzygenowy pomiędzy genami 9R/10R rodziny wielogenowej 505 (MGF),
- Region oznaczony jako ECO2, zlokalizowany pomiędzy genami I329L i I215L.
Podejście to umożliwia łączną analizę wariantów genetycznych obecnych w różnych regionach genomu, obejmujących zarówno krótkie powtórzenia tandemowe (STR), jak i mutacje punktowe (SNP – polimorfizmy pojedynczego nukleotydu), zapewniając znacznie wyższą rozdzielczość niż analiza pojedynczych markerów.
Specyficzna kombinacja wariantów wykrytych w każdym z tych regionów tworzy charakterystyczny „podpis genetyczny” danego wirusa.
Na tej podstawie zdefiniowano dotychczas 28 podgrup genetycznych w obrębie genotypu II krążącego obecnie w Europie. Podgrupa genetyczna 1 odpowiada podstawowemu profilowi genotypu II i jest związana ze szczepem referencyjnym VPPA Georgia 2007/1.
Klasyfikacja ta ma wyłącznie znaczenie epidemiologiczne i służy śledzeniu molekularnemu oraz analizie dróg szerzenia się wirusa. Nie oznacza ona znanych różnic w wirulencji, transmisyjności ani obrazie klinicznym choroby.
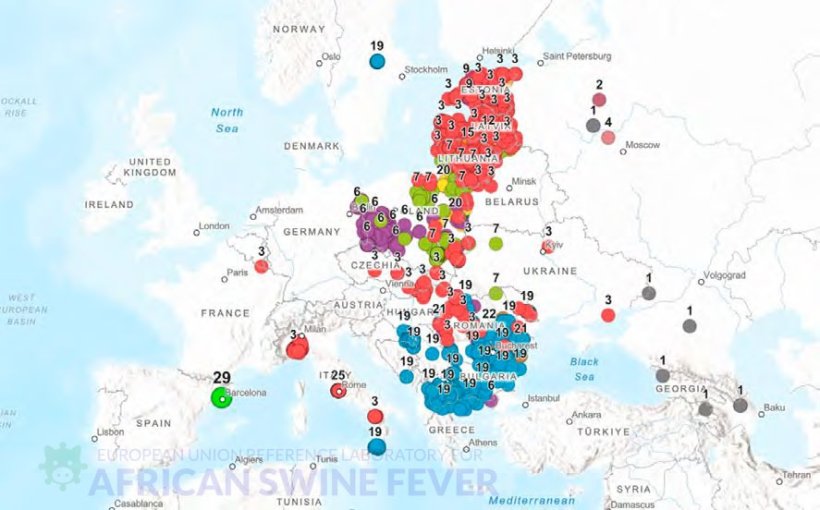
Wirus wykryty u dzików na obszarze Collserola w pobliżu Barcelony wykazał różnice genetyczne w porównaniu z wcześniej opisanymi wirusami. Z tego względu został sklasyfikowany jako przedstawiciel nowej grupy genetycznej – grupy 29 w obrębie genotypu II (Tabela 1).
Tabela 1. Podgrupy genetyczne wirusa ASF genotypu II zdefiniowane metodą wielogenową: profil genetyczny.
| Podgrupa | CVR | IGR I73R / I329L | O174L | K145R | IGR MGF 505 9R/10R | ECO2 |
|---|---|---|---|---|---|---|
| 1 | I | I | I | I | I | I |
| 2 | I | I | I | I | II | I |
| 3 | I | II | I | I | I | I |
| 4 | I | I | I | I | III | I |
| 5 | II | II | I | I | I | I |
| 6 | I | II | II | II | I | I |
| 7 | I | II | I | II | I | I |
| 8 | I | II | I | II | II | I |
| 9 | I-SNP1 | II | I | I | I | I |
| 10 | I | I | II | II | I | I |
| 11 | I-SNP1 | II | I | II | I | I |
| 12 | I | II | I | I | II | I |
| 13 | I | III | II | II | I | I |
| 14 | I-SNP1 | II | I | I | I | I |
| 15 | I | II | I | I | V | I |
| 16 | I | II | I | I | IV | I |
| 17 | I | II | I | I | I-V1 | I |
| 18 | I | III | II | I | I | I |
| 19 | I | II | I | I | I | II |
| 20 | I | IV | I | II | I | I |
| 21 | I | II | II | I | I | I |
| 22 | I | II | II | I | I | II |
| 23 | I | II | I | I | VII | I |
| 24 | I | II | I | I | VI | I |
| 25 | I | II | I | II | VIII | I |
| 26 | I | II | I-SNP1 | I | I | I |
| 27 | I | II | I | II | V | I |
| 28 | I | II | I | I | IX | I |
| 29 | I | II | I | I | I-V2 | I |
Hiszpański izolat wykazuje profil genetyczny blisko spokrewniony z podstawowym profilem genotypu II (Georgia 2007), ponieważ jest zgodny w pięciu spośród sześciu analizowanych regionów, różniąc się jedynie w jednym z nich. W regionie międzygenowym MGF505-9R/10R wykryto mutację (G→A), która nie została wcześniej opisana wśród 1186 wirusów genotypu II znajdujących się w bazie danych EURL-ASF ani w międzynarodowych bazach sekwencji.
Co oznaczają poszczególne warianty?W obrębie każdego analizowanego regionu warianty oznaczane są w sposób ujednolicony:
Dzięki temu możliwe jest rozróżnianie bardzo podobnych wirusów z wysoką dokładnością, przy jednoczesnym zachowaniu spójności pomiędzy poszczególnymi badaniami. |
Ocena pokrycia genetycznego wirusa ASF genotypu II w Europie
Porównywanie nowych ognisk choroby z wcześniej wykrytymi przypadkami wymaga dostępu do aktualnych i reprezentatywnych danych genetycznych dotyczących wirusów krążących w Europie.
Kraje zostały sklasyfikowane na podstawie dostępności danych genetycznych zgromadzonych w bazie sekwencji Europejskiego Laboratorium Referencyjnego ds. ASF oraz oficjalnych zgłoszeń ognisk ASF u świń domowych i dzików.
| Kategoria | Rok ostatniej genetycznie scharakteryzowanej izolacji | Łączna liczba dostępnych zsekwencjonowanych wirusów | Występowanie aktywnej cyrkulacji wirusa ASF | Oficjalne potwierdzenie eradykacji (jeśli dotyczy) | Kraje |
|---|---|---|---|---|---|
| 1 | Do 2025 r. | Reprezentatywna liczba sekwencji | Aktywna cyrkulacja | Estonia, Grecja, Chorwacja, Włochy, Rumunia, Mołdawia | |
| 2 | Aktualne dane genetyczne (2023–2024) | Dostępne są sekwencje związane z ostatnim epizodem epidemiologicznym | Aktywna cyrkulacja lub kraje, w których ASF zostało eradykowane | Belgia, Bułgaria, Czechy, Niemcy, Łotwa, Słowacja, Szwecja, Albania, Bośnia i Hercegowina, Macedonia Północna, Czarnogóra, Kosowo | |
| 3 | Ograniczone dane genetyczne (2021–2022) | Aktywna cyrkulacja | Litwa, Polska | ||
| 4 | Brak danych genetycznych z ostatnich pięciu lat lub bardzo mała liczba dostępnych izolatów | Aktywna cyrkulacja | Węgry, Serbia, Ukraina, Rosja, Armenia, Azerbejdżan, Białoruś, Gruzja | ||
Klasyfikacja ta wykazała istnienie istotnych luk geograficznych i czasowych w dostępnych danych genetycznych dotyczących ASF w Europie.
Tabela 3. Liczba izolatów wirusa ASF genotypu II oraz ich udział procentowy obliczony na podstawie 1187 sekwencji, dla których dostępne były kompletne dane dotyczące sześciu analizowanych regionów genomu.
| Grupa genetyczna | Rozmieszczenie geograficzne (rok) | Liczba | % |
|---|---|---|---|
| 1 | Gruzja (2007), Armenia (2007–2008), Azerbejdżan (2008), Federacja Rosyjska (2009, 2012, 2019), Polska (2022) | 11 | 0.9 |
| 2 | Rosja (2012) | 1 | 0.1 |
| 3 | Ukraina (2012, 2019), Białoruś (2013), Litwa (2014–2018, 2020–2022), Polska (2014, 2018, 2021–2022), Łotwa (2014–2015, 2017–2024), Estonia (2014–2025), Czechy (2017–2018), Rumunia (2017–2019, 2021, 2023–2024), Mołdawia (2017–2018, 2025), Węgry (2018–2019), Belgia (2018), Słowacja (2019–2020, 2022–2023), Włochy (2022–2023), Rosja (2019) | 439 | 37.0 |
| 4 | Rosja (2012) | 1 | 0.1 |
| 5 | Estonia (2015–2016) | 7 | 0.6 |
| 6 | Polska (2016, 2019, 2021–2022), Niemcy (2020–2024), Czechy (2022, 2024) | 154 | 13.0 |
| 7 | Polska (2016–2019, 2021), Litwa (2017–2018, 2020–2022), Rumunia (2019), Łotwa (2021, 2023–2024), Rosja (2018) | 156 | 13.1 |
| 8 | Polska (2016–2017) | 11 | 0.9 |
| 9 | Estonia (2017) | 10 | 0.8 |
| 10 | Polska (2017) | 2 | 0.2 |
| 11 | Polska (2017) | 1 | 0.1 |
| 12 | Łotwa (2017–2018, 2021–2024) | 29 | 2.4 |
| 13 | Polska (2017) | 1 | 0.1 |
| 14 | Litwa (2017), Łotwa (2023–2024) | 4 | 0.3 |
| 15 | Litwa (2017) | 1 | 0.1 |
| 16 | Litwa (2017–2018) | 5 | 0.4 |
| 17 | Łotwa (2017–2018) | 3 | 0.3 |
| 18 | Polska (2018) | 1 | 0.1 |
| 19 |
Rumunia (2018, 2021, 2023–2025), Bułgaria (2018–2020, 2024), Serbia (2019–2020, 2022), Grecja (2020, 2024–2025), Macedonia Północna (2022, 2024), Włochy (2022–2023), Szwecja (2023), Chorwacja (2023–2025), Bośnia i Hercegowina (2023), Czarnogóra (2024), Albania (2024), Kosowo (2024), Mołdawia (2025) |
268 | 22.6 |
| 20 | Polska (2018–2019, 2021–2022) | 23 | 1.9 |
| 21 | Rumunia (2019), Czechy (2024) | 8 | 0.7 |
| 22 | Rumunia (2019) | 12 | 1.0 |
| 23 | Łotwa (2020) | 1 | 0.1 |
| 24 | Rumunia (2021) | 1 | 0.1 |
| 25 | Włochy (Lazio, 2023) | 12 | 1.0 |
| 26 | Włochy (Piedmont, 2023) | 4 | 0.3 |
| 27 | Polska (2021–2022) | 15 | 1.3 |
| 28 | Estonia (2022–2024) | 5 | 0.4 |
| 29 | Hiszpania (2025) | 1 | 0.1 |
Poziom 3. Pełne sekwencjonowanie genomu wirusa wykrytego w Hiszpanii
Pełny genom hiszpańskiego izolatu został poddany sekwencjonowaniu w celu uzyskania najwyższego możliwego poziomu rozdzielczości genetycznej.
Dla szczepu wykrytego w Barcelonie zaproponowano nazwę SP25WB2611, a jego sekwencja została udostępniona w bazie GenBank.
Analiza wykazała, że izolat ten posiada:
- całkowitą długość genomu wynoszącą 180 757 nukleotydów,
- redukcję genomu o około 9,8 kb wynikającą z dużej delecji.
Delecja ta spowodowała całkowitą utratę 21 genów, w szczególności należących do rodzin wielogenowych (MGF), w tym:
- MGF110 (MGF110-7L, -8L, -9L, -10L/14L, -12L, -13La i -13Lb),
- MGF360 (MGF360-4L i MGF360-6L),
- MGF100 (MGF100-1R).
Delecje w tym regionie genomu były wcześniej opisywane zarówno w Europie, jak i w Afryce, choć różniły się zakresem i zawartością genetyczną. Wskazuje to, że tego typu reorganizacja struktury genomu stanowi element naturalnej ewolucji wirusa.
Poza regionem objętym delecją hiszpański izolat wykazuje bardzo wysoką stabilność genetyczną względem szczepu referencyjnego Georgia 2007/1 należącego do genotypu II.
Analiza porównawcza wykazała:
- ponad 99,9% identyczności nukleotydowej we wszystkich wspólnych regionach obu genomów,
- 18 pojedynczych mutacji punktowych (SNP),
- 13 krótkich insercji i delecji (INDEL) o długości poniżej 5 nukleotydów.
Na szczególną uwagę zasługuje delecja obejmująca 5 nukleotydów zlokalizowana w genie należącym do rodziny MGF505. Zmiana ta nie została wcześniej stwierdzona w żadnym z izolatów dostępnych w publicznych bazach danych.
Scenariusze dotyczące pochodzenia wirusa
Dotychczas nie udało się jednoznacznie ustalić konkretnego geograficznego ani epidemiologicznego źródła ogniska. Obecność unikalnej sygnatury genetycznej w hiszpańskim izolacie pozwala wykluczyć jego powiązanie z sekwencjami genotypu II ASFV dostępnymi obecnie w publicznych bazach danych.
Przypadkowe uwolnienie wirusa z laboratorium badawczego
W celu oceny hipotezy o możliwym przypadkowym uwolnieniu wirusa z laboratorium badawczego krajowe laboratorium referencyjne przeanalizowało 81 próbek wirusa wykorzystywanych w badaniach eksperymentalnych prowadzonych w CReSA-IRTA. Żadna z nich nie wykazywała specyficznych markerów genetycznych (SNP) zidentyfikowanych w hiszpańskim izolacie odpowiedzialnym za wystąpienie ogniska.
Uzyskane wyniki nie wykazały zgodności genetycznej pomiędzy hiszpańskim izolatem a wirusami wykorzystywanymi w badaniach eksperymentalnych prowadzonych w CReSA-IRTA, zarówno na poziomie markerów częściowych, jak i całego genomu.
Zawleczenie wirusa z aktywnych ognisk ASF w Europie w wyniku naturalnego lub stopniowego szerzenia się choroby
Scenariusz ten uznano za mało prawdopodobny, ponieważ najbliższe aktywne ognisko ASF w Europie w momencie wykrycia przypadku (we Włoszech) znajdowało się w odległości ponad 500–600 km, a kraje położone pomiędzy tymi obszarami prowadzą skuteczne programy monitoringu ASF. Ponadto profil genetyczny wirusa wykrytego w Hiszpanii nie wykazywał bliskiego pokrewieństwa z wirusami krążącymi we Włoszech.
Celowe wprowadzenie wirusa
W hipotetycznym scenariuszu celowego wprowadzenia wirusa wykorzystanie szczepu podobnego do hiszpańskiego izolatu byłoby mało logiczne, ponieważ charakteryzuje się on wysokim poziomem delecji genomowych oraz niepewnymi właściwościami biologicznymi, co ogranicza możliwość przewidywania jego transmisji i patogenności. Celowe introdukcje są zwykle związane z wykorzystaniem dobrze scharakteryzowanych szczepów o znanych właściwościach epidemiologicznych.
Zawleczenie wirusa na duże odległości za pośrednictwem działalności człowieka
Jest to najczęstszy mechanizm rozprzestrzeniania się wirusa ASF na duże odległości i został szeroko opisany w epidemiologii tej choroby. Scenariusz ten jest zgodny z wieloma elementami epidemiologicznymi zaobserwowanymi podczas ogniska wykrytego w Katalonii:
- Izolowany charakter ogniska.
- Brak ognisk pośrednich w krajach sąsiadujących.
- Lokalizacja ogniska na obszarze o dużej intensywności przemieszczania się ludzi oraz gęstej sieci infrastruktury drogowej i kolejowej.
- Wyraźna odrębność genetyczna względem dominujących linii ASFV krążących w Europie, w tym względem najbliżej spokrewnionych szczepów, co sugeruje zawleczenie niezwiązane ze znanymi szlakami geograficznego szerzenia się choroby.
Charakterystyka obszaru objętego ogniskiem dodatkowo zwiększa prawdopodobieństwo zawleczenia wirusa za pośrednictwem odpadów. Pierwotne ognisko zlokalizowano na terenie, gdzie obszary miejskie sąsiadują z terenami leśnymi i rolniczymi, występują stabilne populacje dzików bez istotnych barier fizycznych, a zwierzęta mają dostęp do pojemników na odpady komunalne, miejsc piknikowych, parków o dużej frekwencji, stref podmiejskich oraz infrastruktury drogowej i kolejowej.
Raport opracowany przez KOMITET NAUKOWY DS. DORADZTWA W ZAKRESIE OGNISKA AFRYKAŃSKIEGO POMORU ŚWIŃ (ASF) W HISZPANII.
Maria del Carmen Gallardo Frontaura
Marta Martínez Avilés
Christian Gortázar Schmidt
Carlos Sánchez García-Abad
Antonio Palomo Yagüe
Daniel Babot Gaspa
Jesús Salas Calvo
Emilio García Muro
Ana Rodríguez Castaño
Niniejszy raport wstępny obejmuje informacje dostępne do dnia 31 stycznia 2026 r.
